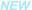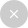Cancer Cell:西湖大学郭天南团队联合荷兰团队发布泛癌蛋白质组图谱
 2025-05-31
2025-05-31
 6651
6651

5 月 29 日,阿姆斯特丹自由大学 Connie R. Jimenez 团队联合西湖大学医学院郭天南团队在 Cancer Cell 在线发表了最新研究,利用数据非依赖性采集质谱(DIA-MS)构建了一个大规模泛癌种蛋白质组图谱(Pan-Cancer Proteome Atlas, TPCPA),覆盖 22 种癌症类型的 999 例原发肿瘤样本,共定量 9,670 种蛋白质。

图 1 文章截图
提纲挈领
研究通过高通量 DIA-MS 技术建立了覆盖 22 种癌症的 Pan-Cancer Proteome Atlas(TPCPA),系统分析了癌种间和同种癌症内的蛋白表达特征,识别出多个泛癌与特异性蛋白标志物、潜在靶点和亚型分类器,特别在结直肠癌的 CMS 亚型与免疫特征分析中展现出良好的临床应用前景,并通过在线平台对外开放了全部数据资源。
作为细胞功能的直接执行者,蛋白质的表达水平比基因组或转录组信息更能反映肿瘤的实际状态。然而,目前大多数癌症研究仍集中在基因层面,缺乏跨癌种的大规模蛋白质组数据资源。
研究利用 DIA-MS 技术构建了涵盖 22 种癌症的高质量蛋白质组图谱,系统解析了不同癌种的蛋白表达特征与生物学功能,一定程度上弥补了这一领域的空白。
通过无监督聚类与加权基因共表达网络分析(WGCNA),研究揭示了癌种间蛋白表达聚类关系,并识别出 13 个功能模块,多个以已知或潜在致癌蛋白为核心,指向肿瘤发生发展的关键分子机制。
同时,针对每个肿瘤排名前 25 的富集蛋白结合转录组、蛋白组、激酶库、血浆可检测性等多种注释信息,优选出了一批具有临床转化潜力的蛋白标志物,包括 MPO(AML)、CEACAM5(结直肠癌)、GFAP(脑癌)等,亦提出了多个新的癌种特异性候选蛋白。
此外,研究在结直肠癌中构建了基于 52 种蛋白的 CMS 亚型分型模型,并开展免疫亚型分析,发现了免疫特征不仅存在于不同癌种之间,也在同种肿瘤内部高度异质。研究还构建了一个 75 蛋白组成的实体肿瘤分类器,在多个外部队列中表现良好,具备对原发灶不明的转移性肿瘤进行原发灶溯源的潜力。
研究最终成果集成至在线数据平台 TPCPA。该平台可查询不同癌种的蛋白表达情况及其功能注释,显著提升了蛋白质组数据的可用性与转化潜力。(http://r2platform.com/TPCPA/)。
以下为研究结果介绍:
01 无监督蛋白质组分析揭示癌种特异性表达模式
研究最终纳入了来自 22 种癌症类型的 999 个高质量原发肿瘤样本,使用 DIA-MS 技术共识别了 11,250 个 protein groups,定量 9,670 种蛋白质,构建了覆盖广泛的泛癌蛋白质组图谱。
降维分析(UMAP)和层次聚类结果显示,样本在蛋白质表达谱上按癌种聚类,且能够有效区分实体瘤与非实体瘤,且不同实验室来源的同癌种样本亦可聚类在一起,说明聚类结果主要反映癌种的分子差异(而非批次效应)。
进一步进行单样本基因富集分析(ssGSEA)重现了多种癌种已知的生物学特征,如前列腺癌中雄激素通路活跃、乳腺癌中雌激素响应增强、肝癌中胆汁酸代谢相关通路富集等。
作为应用实例,研究还挖掘了与 E3 泛素连接酶相关的表达模式,发现多种 E3 酶在特定癌种中高表达(如食管癌中的 HERC5、肝癌中的 RNF5),这些蛋白与多种致癌信号通路和免疫、代谢过程密切相关,提示其有望用于蛋白降解类药物(如 PROTACs)的靶点开发。
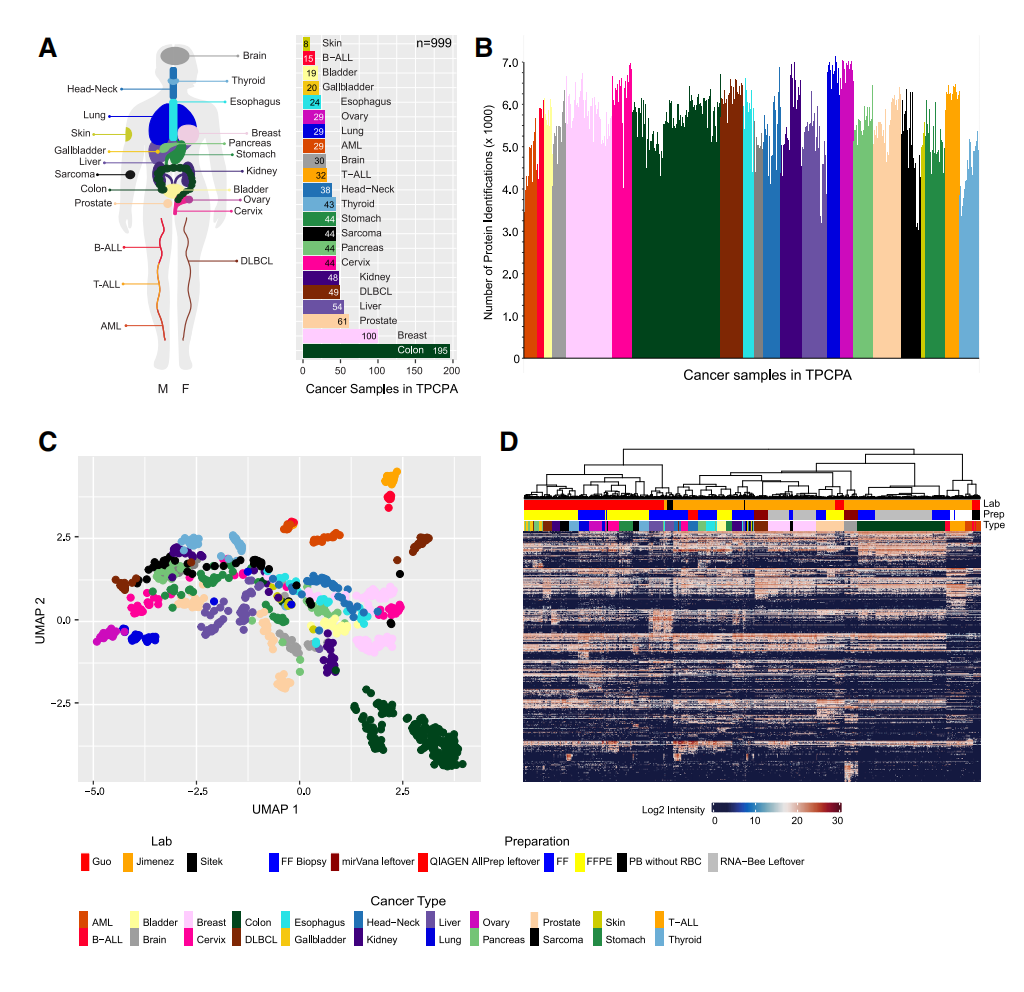
图 2 基于 DIA-MS 构建的泛癌蛋白质蛋白质组景观(The Pan-Cancer Proteome Atlas, TPCPA)
02 共表达网络揭示 13 个癌相关蛋白模块
研究通过 WGCNA 方法共识别出 13 个蛋白协同表达模块,不同模块在各类癌症中呈现特异性表达模式。模块 1–4 主要与细胞黏附和免疫/炎症反应相关,广泛存在于实体瘤中,反映肿瘤微环境的重要作用。模块 5 和 6 则在结直肠癌中显著富集,前者涉及糖基化及细胞连接过程,包含 Wnt 信号通路关键蛋白 β-catenin 和糖胺途径酶 GFPT1/2;后者则关联应激反应,包含热休克蛋白 HSP90AA1/AB1 等潜在药物靶点,并涉及线粒体功能和胞外囊泡分泌。
模块 7 表达范围较广,但与肝组织特征显著相关,主要涉及线粒体代谢与小分子代谢,提示其在多种肿瘤中的基础代谢功能。模块 10 则活跃于鳞状上皮癌(如宫颈癌、头颈癌和食管癌),富含角蛋白等上皮结构蛋白。模块 12 主要在血液肿瘤(如 ALL、DLBCL)中高表达,涉及淋巴器官特征及免疫应答调控,核心蛋白包括 PTPN6、DOCK2、IL16 等,参与 T 细胞迁移、信号转导和免疫调节。
此外,血癌还在模块 9、11、13 中表现出较高活性,这些模块主要与细胞周期、基因表达和抗原呈递等基本生物过程相关。
总体而言,该分析揭示了不同癌种的特异性功能蛋白网络,并识别出一批可能具有转化潜力的新型枢纽蛋白,为靶向治疗提供了候选靶点和生物学基础。
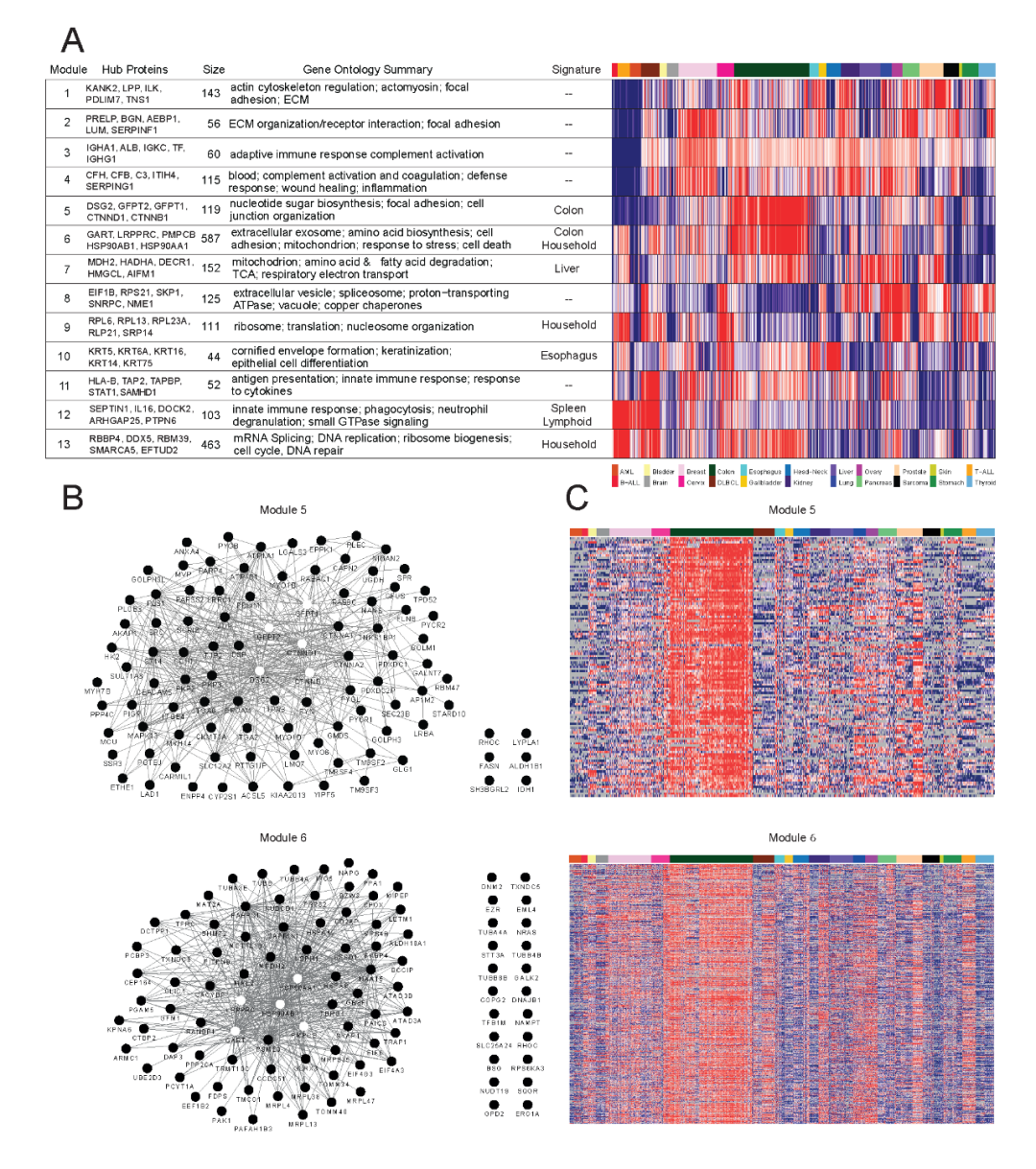
图 3 加权基因共表达网络分析。热图展示结直肠癌相关模块和枢纽蛋白
03 免疫浸润分析揭示肿瘤免疫异质性
ESTIMATE 算法结果显示,非实体肿瘤的免疫评分最高,实体瘤中黑色素瘤呈「免疫热」(immune「hot」)特征,而前列腺、脑和卵巢癌为典型「免疫冷」(immune 「cold」)肿瘤。
ssGSEA 免疫分析揭示癌种间及同种癌内均存在免疫浸润差异。Tamborero 免疫特征在蛋白质组学数据中表现最佳,能够有效区分实体瘤与非实体瘤,识别 ALL 和 DLBCL 中的 T/B 细胞亚型。
结果强调,蛋白质组学数据可以反应肿瘤免疫微环境存在异质性,具有支持肿瘤免疫分析和免疫治疗策略制定的潜力。
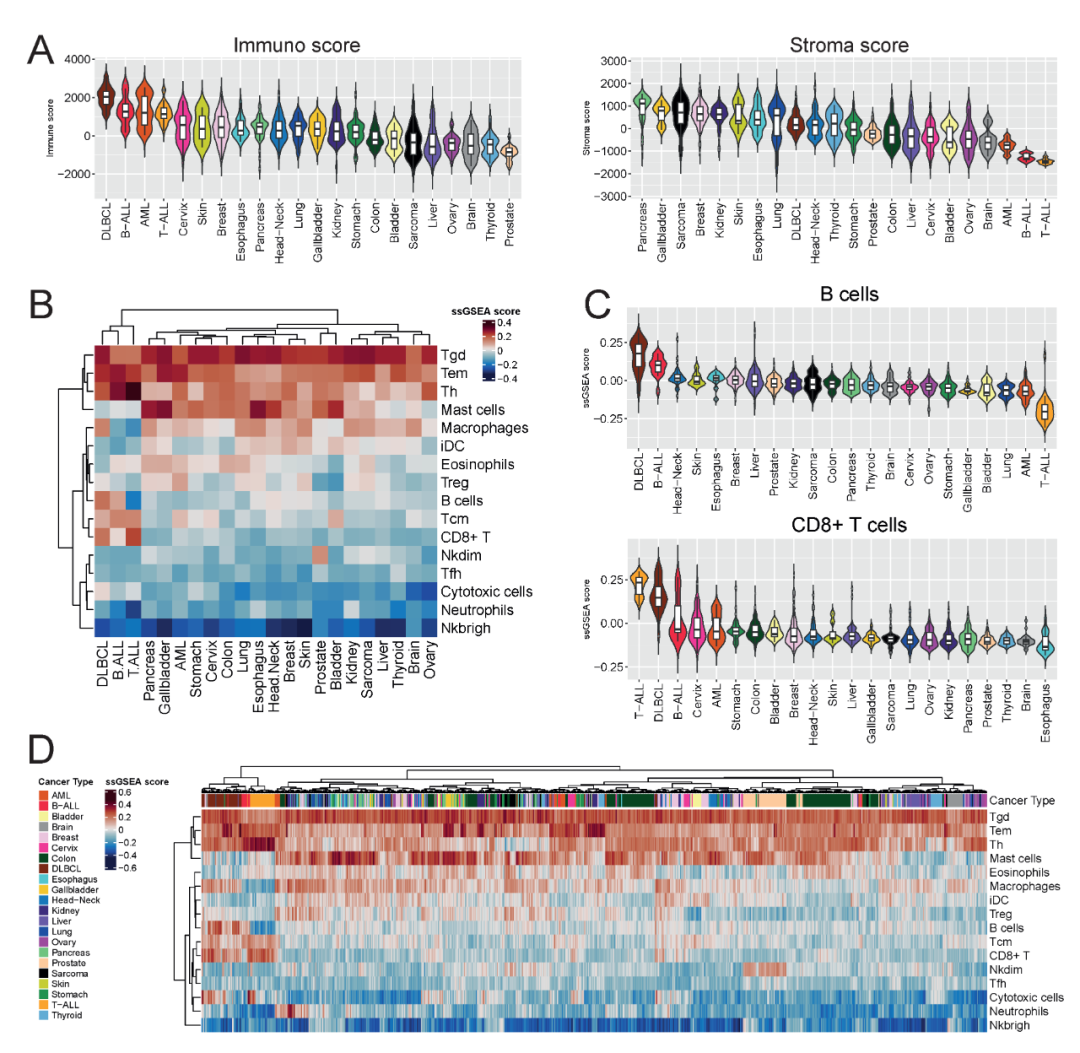
图 4 免疫浸润分析揭示癌症异质性。图中展示 ESTIMATE 评分与免疫亚型聚类结果
04 差异分析揭示泛癌与癌种特异性蛋白,作为潜在靶点和生物标志物
通过差异分析,研究明确了非实体瘤与实体瘤的显著差异蛋白。非实体肿瘤富含免疫和细胞周期相关蛋白(如 BCL7A、IKZF1),实体瘤则富集黏附、应激与迁移相关蛋白(如 EGFR、PTPRK)。这些差异蛋白可作为广谱或泛癌靶点。
癌种间比较发现多数高表达蛋白为已知标志物(如 CEACAM5 用于结直肠癌,TSHR 用于甲状腺癌),也识别出一批此前未在对应癌种中报道的潜在新标志物(如 CAMK2D 在脑癌)。这些蛋白中多数可在血浆中检测,具有无创检测的潜力。
此外,研究人员构建了一个基于 75 个蛋白的实体肿瘤分类器(基于 XGBoost 算法),在内部验证及 4 个独立队列(包括转移癌)中均表现优异(AUC 大于 0.98),表明该方法有望用于对原发灶不明的转移性肿瘤进行原发灶溯源的分析。
05 结直肠癌 CMS 亚型的蛋白组特征及临床关联分析
研究对 191 个结直肠癌样本进行 CMS 亚型蛋白组分析,确认了 4 类 CMS 的生物学特征:CMS1(免疫活跃)、CMS2(增殖)、CMS3(代谢)、CMS4(间充质/免疫抑制/血管生成)。此外,研究人员还富集到此前未被报道的亚型特异性通路,如 CMS1 中 mTORC1 信号通路、CMS4 中氧化应激和低氧通路。
随后,使用亚型特异性蛋白构建一个基于 52 种蛋白的 CMS 亚型分类器,在独立数据集验证中准确率达 72%,表明 DIA-MS 有望在无需转录组信息情况下,使用蛋白质组学数据对结直肠癌的 FFPE 样本进行 CMS 亚型分类。
同时,基于 Tamborero 免疫特征进行共识聚类分析,识别出 3 个免疫亚型(immune CC1–3),并发现它们与 CMS 亚型相关。尤其在 AMC 队列中,富含 CD8+ T 细胞特征的 immune CC1 亚型患者,表现出更长的无复发生存期(RFS)。值得注意的是,基于免疫亚型的预后预测效能显著优于传统 CMS 分子分型,这提示蛋白质组学层面的免疫分型在临床预后评估中具有应用潜力。
06 交互式数据平台 TPCPA
研究搭建了一个功能完备的 TPCPA 数据门户(http://r2platform.com/TPCPA),平台包括五大模块:样本队列总览(Cohort)、蛋白表达可视化(Proteins)、癌种注释与分析(Cancers)、结直肠癌 CMS 亚型特征蛋白/mRNA 表达展示(CRC CMS Protein/mRNA)以及专题分析模块(Dedicated Analyses,涵盖 E3 连接酶及肺癌、乳腺癌的亚型分析)。
使用者可在平台中按癌种、蛋白或分子特征进行灵活查询与交互式可视化,提供开放、实用的资源以促进深入开展癌症蛋白组研究。

图 5 TPCPA 数据门户
总 结
该研究提供全面的基于 DIA-MS 的泛癌蛋白质组资源,不仅填补了转录组与蛋白质表达之间的认知空白,还提供了新的分子视角理解癌症异质性、识别潜在靶点及开发蛋白质水平的癌症分类工具。高通量、可重现的 DIA-MS 平台在 FFPE 样本上的应用也为蛋白质组学生物标志物的临床转化打下技术基础。
研究也存在一些局限性:尽管涵盖多个癌种和实验室,数据整合过程仍可能存在批次效应;某些癌种样本量偏少,限制统计分析的显著性;构建分类器时的交叉验证设计尚有优化空间,存在过拟合风险。因此,研究人员呼吁:未来需在更大规模独立队列中验证研究结果,尤其是蛋白标志物和癌症分类模型的临床应用方面。
西湖大学医学院博士生吕梦葛为本研究共一作者。
论文链接:
https://www.cell.com/cancer-cell/abstract/S1535-6108(25)00212-0