香港科技大学&深圳湾实验室,最新Nature Catalysis!
 2026-02-11
2026-02-11
 19544
19544


研究要点
本研究开发了一种基于α-硫代Rh-卡宾的催化新策略,首次实现了在温和条件下对醇(O–H)和胺(N–H)的高对映选择性插入反应,从而高效构建了结构多样、手性可控的α,α-双杂原子羧酸(图1)。该方法突破了传统金属卡宾化学通常只能引入单一杂原子的局限,通过使用S,S-叶立德作为通用前体、手性质子穿梭催化剂控制立体化学,并结合理论计算阐明了在高度极性的多杂原子环境中实现高对映选择性的机制。所得手性双杂原子砌块不仅结构独特,且可进一步转化为非天然氨基酸(UAAs),并成功应用于肽段修饰和药物分子后期功能化,显著改善了部分抗癌先导化合物的水溶性与活性,为化学合成、药物发现及化学生物学领域提供了强大而通用的新工具。

图1. 催化策略示意图
一、研究背景
手性α-杂原子羧酸是调节多肽、蛋白质及众多生物活性分子结构与功能的关键结构单元。其中,α-氨基酸作为生命的基本构筑单元已广为人知。然而,对于同一碳原子上连接两个不同杂原子的α,α-双杂原子羧酸(DHCAs),其研究尚处于起步阶段。这类结构独特的“非经典”手性中心,尤其是含有两个不同电负性杂原子的情况,能产生独特的非共价相互作用,有望在不显著增加分子量的前提下,有效调节分子的高级结构和功能,在化学生物学、多肽化学与药物研发中潜力巨大(图2)。
然而,其高效合成,特别是对映选择性的控制,一直面临严峻挑战。在同一个碳中心上引入多个杂原子会导致复杂的氢键网络、偶极排列和范德华相互作用,使得反应活性和立体选择性难以预测和控制。金属卡宾介导的不对称X–H键插入反应是构建手性α-杂原子中心的通用策略,但其通常仅能引入一个杂原子。要构建双杂原子手性中心,需要杂原子取代的金属卡宾中间体,而由于电负性杂原子直接连接至卡宾中心会导致前体极不稳定,这类物种的化学长期以来几乎未被探索。
二、研究思路
1. 催化剂设计与反应策略创新
面对多杂原子环境中立体控制难、传统金属卡宾中间体不稳定的双重困境,研究团队提出了一个巧妙的解决方案。他们以易于制备的S,S-叶立德作为通用前体,在Rh(II)催化剂作用下,生成关键的α-硫代Rh-卡宾物种。该策略的核心在于利用硫原子形成氢键倾向较弱的特性。相较于氧或氮,硫的引入可以减少不必要的氢键相互作用,从而促使高度杂原子取代的烯醇中间体主要以单一几何异构体(Z-式)存在。这不仅解决了多杂原子烯醇易产生E/Z异构体混合的问题,同时为后续决定手性的对映选择性质子化步骤提供了至关重要的构象控制基础(图2)。由此,硫原子扮演了“临时杂原子”和“构象控制基团”的双重角色,为最终引入两个不同杂原子铺平了道路。

图2. α-杂原子手性羧酸酯的合成
2. 对映选择性O–H插入反应:构建α,α-O,S-羧酸酯
研究首先聚焦于醇的O–H键插入。经过系统优化,确定了以手性Rh₂(S-TCPTTL)₄配合物与Takemoto手性氨基硫脲衍生物(CAT-1)作为最优催化组合。该体系能够在温和条件(40°C,DCM)下,高效、高对映选择性地实现一系列S,S-叶立德与各类醇的反应(图3)。反应展现出广泛的底物适用范围和官能团耐受性:芳硫基部分可兼容富电子或缺电子取代基、萘环、噻吩等;酯基部分可灵活变换为不同链长或分支的烷基、烯丙基等;醇组分则涵盖伯醇、仲醇及含有NHBoc、环丙烷、硅基、烯烃等多种官能团的复杂分子。值得注意的是,反应具有优异的化学选择性,空间位阻最小的羟基优先反应。该工艺可轻松放大至克级规模而不影响产率和对映选择性。

图3. α,α-O,S-羧酸酯的底物范围
3. 对映选择性N–H插入反应:通向α-硫代氨基酸
在O–H插入成功的基础上,研究进一步拓展至N–H键插入,以直接获取手性α-硫代氨基酸——一类重要的非天然氨基酸。优化发现,对于N–H插入,手性螺环磷酸(CPA-9) 的表现优于硫脲类催化剂,能实现更高的反应活性和对映选择性。N–H插入的底物范围比O–H插入更广,伯、仲、叔烷基氨基甲酸酯均能兼容,以良好至优秀的收率和对映选择性得到目标产物(图3)。芳硫基片段上的卤素、醚、醛、氰基、羰基等官能团对反应影响甚微。尤为突出的是,对于O–H插入表现一般的伯烷硫基底物,在N–H插入中反而表现出更好的性能。此外,从松香醇、L-薄荷醇、吉非罗齐等复杂天然产物或药物分子衍生的片段也能成功接入,展示了该方法的后期官能化潜力。

图4. α,α-N,S-羧酸酯的底物范围
4. 理论计算揭示立体控制根源
为了深入理解在多杂原子环境下实现高对映选择性的机理,团队进行了系统的密度泛函理论计算(图5)。计算明确了反应经由Z-式烯醇中间体进行,其在动力学和热力学上均更有利。对于决定对映选择性的质子化步骤,计算比较了从Z-和E-式烯醇出发产生R型和S型产物的所有可能过渡态。结果表明,从Z-式烯醇形成R-产物的过渡态能量最低,这与实验观测到的绝对构型一致。进一步的扭曲/相互作用分析和AIM分析揭示,在优势过渡态中,手性磷酸催化剂(CPA-9)的3,3'-位均三甲苯基与烯醇中间体的NBoc基团之间形成了更多、更强的非共价相互作用(如C–H···H–C、π···孤对电子(S)、N–H···π作用等)。这些相互作用网络像“分子手套”一样,更好地识别并稳定了生成R-构型产物的过渡态,从而实现了优异的立体控制。
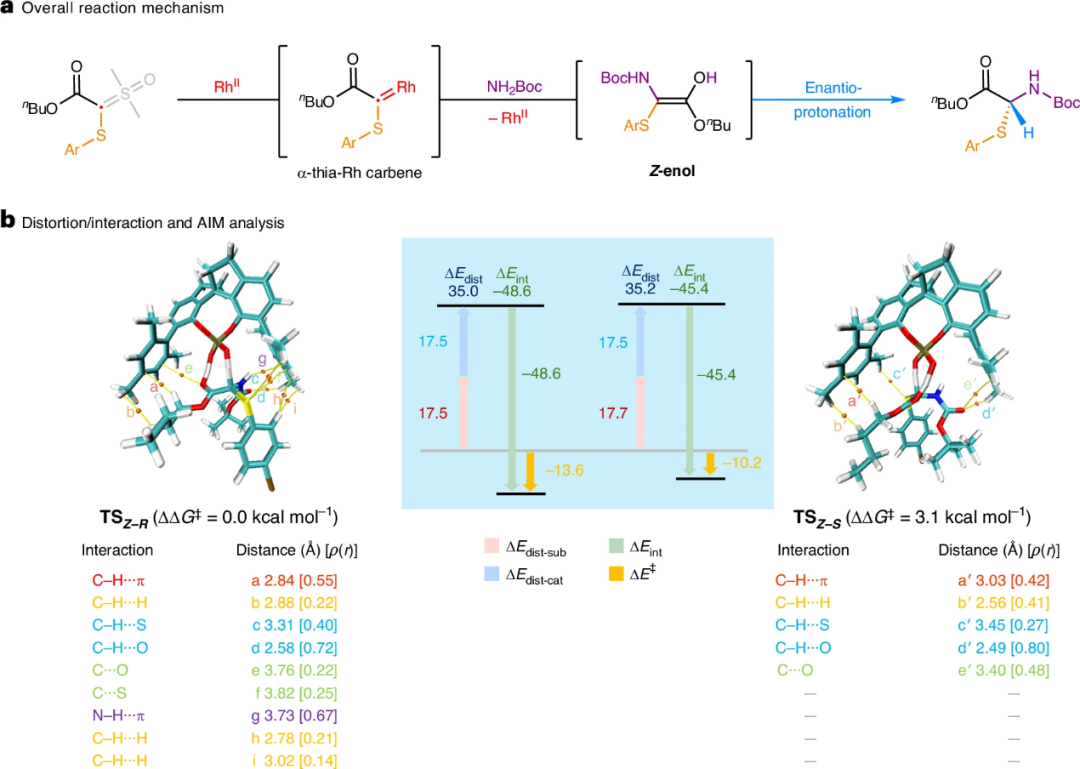
图5. 关键过渡态的计算分析
5. 合成应用与功能拓展
构建多样化的α-杂原子非天然氨基酸库
所得的手性α,α-双杂原子羧酸酯是极佳的多功能合成砌块。研究发展了一种硫原子交换策略,能够将对映选择性地将α-硫原子替换为氧或氮等其他杂原子,从而形式上实现了“双X–H键插入”,获得了一系列之前难以获取的α-氧基和α-氨基非天然氨基酸(图6a-c)。这些砌块可方便地转化为羧酸、Weinreb酰胺、酰胺、酮、炔酮、醇等多种官能团(图6d),连通性极强。

图6. 合成应用
化学空间分析与性质评估
通过t-SNE和PCA等数据降维可视化分析,本工作合成的31种α-杂原子非天然氨基酸在结构上形成了一个独特的“岛屿链”,与现有的天然氨基酸及常见非天然氨基酸化学空间形成互补,同时又与它们在整体理化性质上保持较高相似性(图7)。这意味着将这些砌块引入生物大分子时,有望在引起显著结构变化的同时,仅带来适中的理化性质改变,这对理性设计生物活性分子具有重要意义。稳定性测试表明,不同类型的双杂原子羧酸酯在酸/碱条件下的稳定性及外消旋化倾向各异,为其在不同场景下的应用提供了指导。

图7. t-SNE与PCA在已知氨基酸化学空间中对α-杂环氨基酸残基的可视化分析
在肽合成与药物分子修饰中的应用展示
研究证明了这些α-杂原子非天然氨基酸能顺利用于肽键合成,与多种天然氨基酸(如Phe, Met, Trp, Ala等)偶联均可获得优异的非对映选择性(图8)。更重要的是,该方法被成功用于六种具有复杂结构的药物分子的后期多样化修饰,包括用于中枢神经系统疾病的缬苯那嗪、舍曲林,用于糖尿病的西格列汀,以及抗癌药物依喜替康、喜树碱、阿霉素等(图8b)。偶联过程在保持α-双杂原子手性中心立体化学完整性的同时,取得了良好至优秀的产率。
尤为引人注目的是,通过引入α-乙氧基非天然氨基酸对喜树碱和依喜替康骨架进行修饰,显著改善了其水溶性。与引入天然氨基酸(甘氨酸、苯丙氨酸)的类似物相比,α-乙氧基修饰的类似物溶解度提升了45倍以上(图8c)。细胞毒性测试进一步显示,α-乙氧基修饰的类似物在多种人癌细胞系中表现出更强、更广谱的抑制活性(IC50普遍低于200 nM),其效力相较于对应的甘氨酸或苯丙氨酸衍生物提升可达5-10倍。这一结果挑战了传统的构效关系认知:在α-中心引入极性杂原子通常被认为可能降低膜渗透性,但本研究表明,通过精心设计,这种修饰反而可以增强靶点亲和力,并有望克服由传统骨架引起的耐药性。

图8. 肽类与药物分子的后期修饰
三、小结
本研究发展了一种普适性强、可编程的新方法,通过对α-硫代Rh-卡宾进行高对映选择性的O–H和N–H键插入,成功解锁了具有独特电子和结构性质的α,α-双杂原子羧酸这一全新的手性分子库。关键在于利用硫的独特性质稳定关键中间体,并借助手性质子穿梭催化剂在多杂原子极化环境中实现精准的立体控制。
该工作不仅拓展了不对称插入反应的范围,更通过合成一系列结构新颖的α-杂原子非天然氨基酸,并将其成功应用于多肽和药物分子的功能化,展示了其在合成化学、多肽化学、化学生物学及药物发现领域的巨大应用潜力。所揭示的“硫导向-质子穿梭控制”策略为在碳中心对映选择性引入两个杂原子提供了通用方案,也为复杂生物分子的构建与修饰提供了宝贵的新工具。未来,通过进一步拓展杂原子类型、优化催化剂体系、并将其应用于更多样化的生物活性分子工程中,这一平台技术有望催生更多功能独特的创新分子。
原文详情:
Xing, Y., Fang, Y., Zhao, Y. et al. Harnessing thia-Rh-carbenes for the enantioselective synthesis of chiral α,α-diheteroatomic carboxylic acids. Nat. Catal. (2026).